

Von Sonia Elijah / Trial Site News
![]()
Im Juni veröffentlichte Trial Site News einen bahnbrechenden Enthüllungsbericht über die geleakten E-Mails der Europäischen Arzneimittelbehörde (EMA) und andere vertrauliche Berichte von Pfizer, die wichtige Fakten im Vorfeld der Zulassung des Corona-Impfstoffs von Pfizer-BioNTech enthüllten. Dieser Bericht deckte auf:
![]()
- Ein politisch motivierter Wettlauf zwischen den wichtigsten Regulierungsbehörden bei der Zulassung des Impfstoffs.
- Ende November 2020 wussten die Aufsichtsbehörden, darunter die US-amerikanische FDA, die Europäische Arzneimittelagentur (EMA), Health Canada und die britische MHRA, vom erheblichen Verlust der RNA-Integrität der kommerziellen Chargen (~55% mRNA-Integrität) des Impfstoffs von Pfizer-BioNTech im Vergleich zu den klinischen Chargen (~78% mRNA-Integrität). Dies wurde von der EMA zusammen mit den beobachteten sichtbaren Partikeln, die als “Verunreinigungen” eingestuft wurden, als “wichtiger Einwand” eingestuft.
- Eine am 26. November geleakte PowerPoint-Präsentation eines Treffens zwischen Pfizer-BioNTech und der EMA offenbarte, wie dieser schwerwiegende Einwand auf schockierende Weise “gelöst” wurde – die Spezifikation für die RNA-Integrität wurde einfach auf 50% gesenkt, d. h. die Hälfte aller mRNA-Moleküle in den kommerziellen Chargen durften abgeschnitten (nicht intakt) sein.
- Die möglichen Auswirkungen des Verlusts der RNA-Integrität auf die Sicherheit und Wirksamkeit waren unbekannt.
Dieser Artikel befasst sich mit weiteren geleakten E-Mails, insbesondere mit der EU-Kommissarin Ursula von der Leyen und dem ungewöhnlichen Ausmaß, in dem sie bereit war, die Mitgliedstaaten dazu zu bringen, die Anwendung von Artikel 5 (2) (ihre nationale Notfallzulassung für die Corona-Impfstoffe) zu vermeiden und stattdessen eine EU-Zulassung für das bedingte Inverkehrbringen (CMA) zu beantragen. Außerdem werden weitere geleakte sensible Dokumente der EMA beleuchtet: die Präsentation der Beobachtungen des Quality Office CMC vom 24. November 2020 durch die BWP (Biologics Working Party) und der Bewertungsbericht des Berichterstatters für die turnusmäßige Überprüfung, der weitere Beweise für die im ursprünglichen Bericht von Trial Site News erörterten “wesentlichen Einwände” enthält. Es handelt sich um ungeschwärzte Fassungen von Pfizer-Impfstoffverträgen und die im November 2020 mit Pfizer und BioNTech unterzeichnete Vorabkaufvereinbarung (Advance Purchase Agreement, APA) “für die Entwicklung, Produktion, vorrangige Kaufoptionen und Lieferung eines erfolgreichen Corona-Impfstoffs für die EU-Mitgliedstaaten”.
![]()
Skandale sind ihr nicht fremd
Ursula von der Leyen stand unter intensiver öffentlicher Beobachtung, weil sie bei den privaten Verhandlungen über ein milliardenschweres Impfstoffgeschäft (den größten Vertrag der EU) geheime Texte und Telefonate mit dem Vorstandsvorsitzenden von Pfizer, Albert Bourla, führte und damit gegen den Beschluss der Kommission verstieß, einen Lenkungsausschuss einzurichten, der “während des gesamten Bewertungsprozesses beratend tätig sein sollte”. Außerdem weigerte sie sich, der Öffentlichkeit Zugang zu den geheimen Textnachrichten zu gewähren, die zwischen Bourla und ihr ausgetauscht wurden, als sie dazu aufgefordert wurde. Der Europäische Rechnungshof veröffentlichte einen alarmierenden Bericht, in dem es heißt:
![]()
“Wir haben die Kommission gebeten, uns Informationen über die Vorverhandlungen zu diesem Abkommen zu übermitteln (konsultierte wissenschaftliche Sachverständige und erhaltene Ratschläge, Zeitplan der Gespräche, Aufzeichnungen der Gespräche und Einzelheiten zu den vereinbarten Bedingungen). Es wurden jedoch keine Informationen vorgelegt.“
Auch für Bourla von Pfizer wird es immer brenzliger, denn er sollte vor dem COVID-19-Sonderausschuss des Europäischen Parlaments aussagen, um sich Fragen zu den geheimen Impfstoffgeschäften zu stellen, sagte aber in letzter Minute ab.
![]()
Die geleakten E-Mails
Der ursprüngliche Enthüllungsbericht von Trial Site News machte den enormen Druck deutlich, den die Europäische Kommission auf die EMA ausübte, um die CMA in einem stark beschleunigten Zeitrahmen zu erteilen. Eine E-Mail von Noel Wathion (ehemaliger stellvertretender Exekutivdirektor der EMA) wurde veröffentlicht, die eine “ziemlich angespannte” Telefonkonferenz mit der EU-Kommissarin (Ursula von der Leyen) enthüllte, die “zeitweise sogar etwas unangenehm” war.
![]()
“Eine Verzögerung von mehreren Wochen war für die EK [Europäische Kommission] nicht ohne weiteres akzeptabel”, so Wathion. Er zeigt auch auf, “dass die politischen Auswirkungen zu groß zu sein scheinen, selbst wenn die ‘technische’ Ebene in den Mitgliedstaaten eine solche ‘Verzögerung’ rechtfertigen könnte, um das Ergebnis der wissenschaftlichen Überprüfung so solide wie möglich zu gestalten”.
![]()
Nachstehend eine E-Mail von Hilde Boone von der EMA, aus der hervorgeht, dass “sie [von der Leyen] bereit ist, die zuständigen Gesundheitsminister persönlich anzurufen, um die Anwendung von Artikel 5 Absatz 2 zu vermeiden”.
![]()

Artikel 5 (2) versus CMA
In Artikel 5 (2) der Richtlinie 2001/83 heißt es:
![]()
“Die Mitgliedstaaten können als Reaktion auf die vermutete oder bestätigte Ausbreitung von krankheitsverursachenden Agenzien, Toxinen, chemischen Stoffen oder schädlicher nuklearer Strahlung vorübergehend das Inverkehrbringen eines nicht zugelassenen Arzneimittels genehmigen.”
Mit anderen Worten: Es handelt sich um das Äquivalent einer Notfallgenehmigung auf der Ebene der Mitgliedsstaaten. Sie kann sehr schnell erteilt werden, da das Arzneimittel nicht das übliche nationale Zulassungsverfahren durchlaufen muss.
![]()
Ein Experte für EU-Recht erklärte Trial Site News, dass der Nachteil der Inanspruchnahme von Artikel 5 (2) ist, dass dies zu einem Wettbewerb zwischen den Mitgliedstaaten geführt hätte, was einen ungleichen Zugang/Verteilung der Corona-Impfstoffe zur Folge gehabt hätte, insbesondere für diejenigen Mitgliedstaaten, die es vorzogen, auf die EU-Zulassungsbehörde zu warten (was länger dauert, da sie einem geregelten und soliden Rahmen folgen soll, der Schutzmaßnahmen vorsieht). Eine EU-Zulassungsgenehmigung trug dazu bei, dass die Impfstoffe allen Mitgliedstaaten zur gleichen Zeit zur Verfügung standen, was eines der Ziele der COVID-19-Strategie der EU war.
![]()
Könnte es jedoch noch weitere Gründe gegeben haben, warum von der Leyen unbedingt wollte, dass die Mitgliedstaaten Art. 5 (2) umgehen, so dass sie bereit war, jeden zuständigen Gesundheitsminister selbst anzurufen?
![]()
In der nachstehenden E-Mail von Noel Wathion heißt es: “Da die erste Option immer noch darin besteht, eine CMA anzustreben und nicht den Artikel 5 (2) in Verbindung mit oder ohne Artikel 5 (3)…”.
![]()

In Artikel 5 Absatz 3 heißt es:
![]()
“Die Mitgliedstaaten erlassen Vorschriften, um sicherzustellen, dass die Inhaber der Genehmigung für das Inverkehrbringen, die Hersteller und die Angehörigen der Gesundheitsberufe nicht zivilrechtlich oder verwaltungsrechtlich für die Folgen der Verwendung eines Arzneimittels haftbar gemacht werden können…”
Die Tatsache, dass Wathion darauf hinweist, dass Art. 5 (2) mit oder ohne Art. 5 (3) verbunden sein kann, wirft die wichtige Frage auf, ob die Mitgliedstaaten die Möglichkeit gehabt hätten, den Zulassungsinhabern (in diesem Fall BioNTech) und den Herstellern (BioNTech und Pfizer) keine Haftungsfreistellung zu gewähren.
![]()
Die betrügerischen Pfizer-Verträge und Vorabkaufvereinbarungen
In Anbetracht dessen, was wir über die an die Öffentlichkeit gelangten Pfizer-Verträge wissen, die von der gemeinnützigen Verbraucherschutzorganisation Public Citizen zur Verfügung gestellt wurden, hat dieses Pharmaunternehmen anscheinend Länder zum Schweigen gebracht; es kann auf das Vermögen souveräner Staaten zugreifen (im Rahmen der Aufhebung der souveränen Immunität) und genießt volle Haftungsfreistellung, d. h. Befreiung von der gesetzlichen Haftung, die sich aus seinem Produkt ergeben kann – in Wirklichkeit ist es der Käufer, der in ihrem Namen haftbar gemacht wird. Dies bedeutet, dass die Regierungen Entschädigungen an Bürger zahlen müssen, die unter den Nebenwirkungen eines Impfstoffs gelitten haben, und nicht der Impfstoffhersteller.
![]()
Der nachstehende Screenshot stammt aus der ungeschwärzten Fassung des Vorabkaufvertrags (APA) zwischen der im Namen und im Auftrag der Mitgliedstaaten handelnden Europäischen Kommission (EK), Pfizer Inc. und BioNTech (zusammen “der Auftragnehmer”), der im November 2020 unterzeichnet wurde (etwa zur gleichen Zeit, als die geleakten EMA-E-Mails geschrieben wurden). Darin heißt es:
![]()
“jeder teilnehmende Mitgliedstaat den Auftragnehmer und seine verbundenen Unternehmen … von jeglicher Haftung … für Schäden und Verluste …, die sich aus der Verwendung und dem Einsatz der Impfstoffe ergeben oder damit zusammenhängen, freistellt und schadlos hält“.

Auf der Internetseite der Europäischen Kommission heißt es jedoch, dass im Rahmen einer bedingten EU-Zulassung für das Inverkehrbringen (CMA) “der Inhaber der Zulassung haftet” (in diesem Fall BioNTech). Dies steht in direktem Widerspruch zu der APA, die die Europäische Kommission einen Monat vor Erteilung der CMA mit Pfizer und BioNTech unterzeichnet hat (für 200 Millionen Impfstoffdosen zu einem Preis von 15,50 Euro ohne MwSt. pro Dosis). Es ist bemerkenswert, dass die Europäische Kommission eine stark geschwärzte Version der identischen APA veröffentlicht hat; alle Abschnitte, die sich auf Entschädigung/Haftung beziehen oder einfach als “sensibel” gelten, wurden zensiert.
![]()
Ein weiterer wichtiger Punkt, der bei der Gegenüberstellung von Artikel 5 Absatz 2 und einer EU-Zulassungsgenehmigung zu berücksichtigen ist, ist die Tatsache, dass einige europäische Länder den Impfstoff für Erwachsene, Risikogruppen und bestimmte Berufsgruppen vorgeschrieben haben – es ist höchst unwahrscheinlich, dass diese Mitgliedstaaten in der Lage gewesen wären, dies mit einem nicht zugelassenen Arzneimittel zu tun, da diese Impfstoffe gemäß Artikel 5 Absatz 2 eingestuft worden wären.
![]()
Die CMC-Problematik
In Wathions E-Mail vom 20. November 2020 werden mehrere besorgniserregende Punkte angesprochen:
![]()
“Es gibt immer noch Probleme mit beiden (CMC scheint ein Problem für Pfizer/BioNTech zu sein, und die rollierende Überprüfung für Moderna hat gerade begonnen, so dass weniger Zeit für die Überprüfung zur Verfügung steht), so dass man abwarten muss, ob all dies rechtzeitig geklärt werden kann, ohne die Zuverlässigkeit der Überprüfung zu gefährden.”
Im ursprünglichen Bericht von Trial Site News wurden die CMC-Probleme (Chemistry Manufacturing and Controls) mit Pfizer/BioNTech erörtert, insbesondere der Verlust der RNA-Integrität in den kommerziellen Chargen und die unbekannten sichtbaren Partikel, die entdeckt wurden. Die Aussage von Wathion, dass es “immer noch Probleme gibt”, deutet darauf hin, dass diese “Probleme” nicht gelöst sind, sondern weiter bestehen. Wathions Sorge, dass die “Zuverlässigkeit der Überprüfung ” beeinträchtigt werden könnte, weil die Genehmigung “rechtzeitig” erteilt werden muss, wird hervorgehoben. Diese Sorge, dass Geschwindigkeit vor Sicherheit geht, spiegelt sich auch in anderen geleakten E-Mails wider, insbesondere in denen von Wathion.
![]()
Der Bericht enthielt auch eine geleakte E-Mail von Veronika Jekerle, der Leiterin des EMA-Büros für pharmazeutische Qualität, in der sie die drei vereinbarten Haupteinwände und die Schlussfolgerung der BWP (Biologics Working Party) bezüglich des Impfstoffs von Pfizer-BioNTech darlegte. Ihre E-Mail wurde am 24. November, dem gleichen Tag wie die BWP-Präsentation, versandt. Nachstehend eine Reihe von Screenshots der geleakten BWP-Powerpoint-Präsentation (auf die sich Jekerle in ihrer E-Mail bezieht) mit dem Titel “EMA Quality Office CMC Observations”. Am Ende ihrer E-Mail dankt sie “Ton, Brian und Claudio”. Ton van der Stappen und Brian Dooley sind im Screenshot der Folie unten genannt.
![]()

Der nachstehende Screenshot zeigt umfangreiche Daten, die einen der wichtigsten Einwände der EMA in Bezug auf den erheblichen Rückgang der prozentualen RNA-Integrität zwischen den klinischen und den kommerziellen Chargen untermauern. Es ist bemerkenswert, dass Chargen aus den Pfizer-Standorten Andover, USA (~62%) und dem belgischen Puurs (~55%) eine deutlich niedrigere prozentuale RNA-Integrität aufwiesen als Chargen von BNT (BioNTech) und Polymun.
![]()
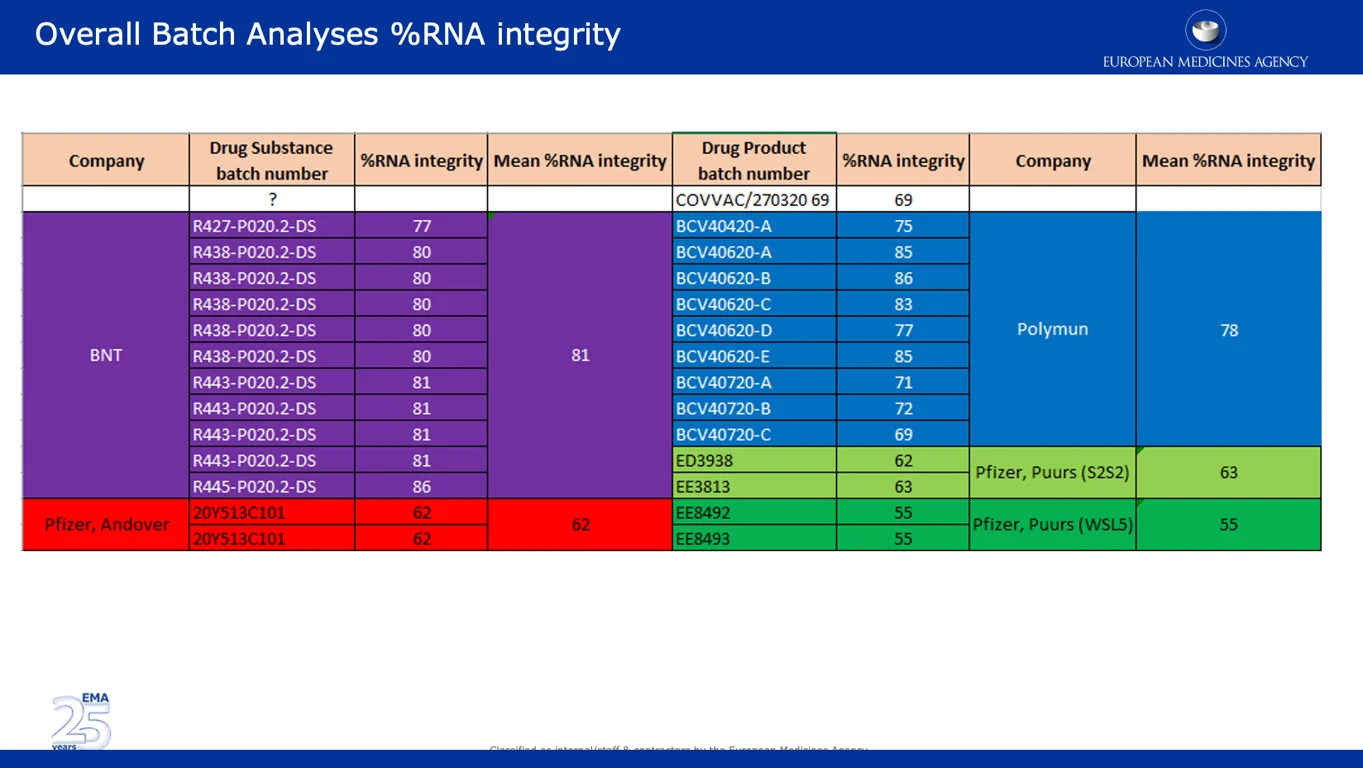
Der folgende Screenshot stammt aus dem von der Europäischen Kommission unterzeichneten ungeschwärzten APA. Darin heißt es, dass der größte Teil der europäischen Impfstofflieferungen “von der Pfizer-Produktionsstätte in Puurs, Belgien” stammen wird. Dies ist besorgniserregend, da die Daten zeigen, dass die prozentuale RNA-Integrität der Chargen von diesem Standort mit 55% im Vergleich zu anderen Standorten am niedrigsten war.
![]()

Die nachstehende Abbildung zeigt die Akzeptanzkriterien für verschiedene Chargen des BNT 162B2 Drug Product. Der Begriff “Drug Product” bezieht sich auf “Arzneimittel in ihrer vermarkteten Form, einschließlich ihrer Füllstoffe, Farbstoffe und anderer aktiver oder inaktiver Wirkstoffe”. Das Akzeptanzkriterium für die Notfallversorgung ist größer oder gleich 50%, was zufällig knapp unter den Chargen mit der niedrigsten prozentualen RNA-Integrität liegt, die von Pfizer in Puurs geliefert wurden. Aus irgendeinem Grund sind die Akzeptanzkriterien für die klinischen Arzneimittelchargen anders und höher angesetzt, nämlich größer oder gleich 60%.
![]()

CMC (Chemistry, Manufacturing and Controls) “umfasst die Festlegung von Herstellungspraktiken und Produktspezifikationen, die befolgt und eingehalten werden müssen, um die Produktsicherheit und die Konsistenz zwischen den Chargen zu gewährleisten.” Angesichts der oben gezeigten Daten ist es offensichtlich, dass es erhebliche CMC-Probleme in Bezug auf die Inkonsistenz der prozentualen RNA-Integrität zwischen den einzelnen Chargen gab, was sich in den geleakten EMA-E-Mails und Dokumenten widerspiegelt. Besorgniserregend ist, dass der Hersteller (Pfizer/BioNTech) behauptete: “Die Wirksamkeit des Arzneimittels hängt von der Expression der zugeführten RNA ab, die ein ausreichend intaktes RNA-Molekül erfordert.”
![]()
Es ist verblüffend, wie durch die Herabsetzung der Spezifikation auf 50% dieser wichtige Einwand möglicherweise “gelöst” wurde. Laut einem geleakten “Rapporteur’s Rolling Review Assessment” (überarbeitetes Berichtsdatum: 25. November 2020), das von den CHMP- und PRAC-Berichterstattern erstellt wurde, fanden sie die “aktuellen Akzeptanzkriterien” besonders beunruhigend. In dem Bericht heißt es:
![]()
“Es werden keine Charakterisierungsdaten zur RNA-Integrität und zu verkürzten Formen vorgelegt, und die potenziellen Sicherheitsrisiken im Zusammenhang mit verkürzten RNA-Isoformen werden nicht angesprochen. Dies ist besonders wichtig, wenn man bedenkt, dass die derzeitigen DS- und DP-Akzeptanzkriterien bis zu 50% fragmentierte Spezies zulassen.”

Abgeschnittene (gekürzte, da entweder der obere oder der untere Teil fehlt) RNA wird als “produktbezogene Verunreinigung” definiert, und die Tatsache, dass potenzielle Sicherheitsrisiken, die von dieser fragmentierten Spezies ausgehen, “nicht berücksichtigt werden”, ist höchst alarmierend.
![]()
Es wird auf “die aktuellen DS- und DP-Akzeptanzkriterien” verwiesen, was bedeutet, dass diese nicht immer auf diesem Niveau lagen und vielleicht geändert (herabgesetzt) wurden. Die Frage ist, warum? Könnte es sein, dass Prozess 2 (die Herstellung der kommerziellen Chargen) schlichtweg nicht auf demselben Spezifikationsniveau replizierbar war wie die klinischen Chargen (im kleinen Maßstab) von Prozess 1, weshalb ein niedrigerer Standard festgelegt wurde, damit die CMA erteilt werden konnte?
![]()
Trial Site News hat sich mit der EMA über den Inhalt der geleakten E-Mails und Dokumente in Verbindung gesetzt. Die prompte Antwort der EMA-Pressestelle wird nachstehend in vollem Wortlaut veröffentlicht.
![]()
“Die Untersuchung des veröffentlichten Materials ergab, dass die Korrespondenz von dem oder den Tätern vor der Veröffentlichung manipuliert wurde. Nicht alle Dokumente wurden in ihrer vollständigen, ursprünglichen Form veröffentlicht und sind möglicherweise aus dem Zusammenhang gerissen worden. Während einzelne E-Mails authentisch waren, wurden Daten von verschiedenen Nutzern ausgewählt und zusammengefasst, Screenshots von mehreren Ordnern und Postfächern erstellt und von den Tätern zusätzliche Titel hinzugefügt.
Diese Dokumente geben kein vollständiges Bild der Bewertung von Comirnaty, dem von BioNTech/Pfizer entwickelten Corona-Impfstoff, wieder. Sie zeigen die Situation bis Anfang Dezember 2020, als der Hack entdeckt wurde, erwähnen aber nicht die beträchtliche Menge an zusätzlichen Daten, Informationen und Klarstellungen, die BioNTech/Pfizer bis zum 21. Dezember 2020, also bis zu dem Tag, an dem der Ausschuss für Humanarzneimittel (CHMP) der EMA seine Empfehlung zur Erteilung einer Zulassung für diesen Impfstoff abgab, vorgelegt hat.
Comirnaty wirkt, weil die darin enthaltene mRNA Anweisungen für die Produktion eines Spike-Proteins liefert, das eine Immunreaktion auslöst. Seine Wirksamkeit hängt daher vom Vorhandensein einer geeigneten Menge intakter mRNA ab, die bekanntermaßen relativ instabil ist. Die Dokumente zeigen, wie die Bewertung eines jeden Arzneimittels abläuft: Nach der Prüfung der vom Unternehmen vorgelegten Daten hatte der CHMP Fragen zur Integrität der mRNA und erhob diese formell als ‘wichtigen Einwand’. Dies ist ein fester Bestandteil der Bewertung eines jeden Arzneimittels. Bleiben wichtige Beanstandungen ungelöst, schließen sie die Erteilung der Genehmigung für das Inverkehrbringen aus. In diesem Fall hat das Unternehmen die erhobenen Einwände zufriedenstellend gelöst und die erforderlichen Informationen und Daten Anfang Dezember 2020 nachgereicht, so dass die EMA eine positive Beurteilung für diesen Impfstoff abgeben konnte.
Der öffentliche Beurteilungsbericht von Comirnaty fasst die Schlussfolgerungen des CHMP zu diesem Thema zusammen und beschreibt die Schritte, die während des Zulassungsverfahrens von Comirnaty unternommen wurden, sowie die Verpflichtungen, die dem Zulassungsinhaber auferlegt wurden, zusätzliche Studien durchzuführen, um die pharmazeutische Qualität des Impfstoffs genau zu überwachen. Diese Verpflichtungen wurden auch in die Produktinformation aufgenommen, die zum Zeitpunkt des CHMP-Gutachtens veröffentlicht wurde.
Selbst bei einem Notfall im Bereich der öffentlichen Gesundheit wie COVID-19 bestand in der EU immer ein Konsens darüber, keine Kompromisse einzugehen und jede Empfehlung auf die verfügbaren wissenschaftlichen Beweise für die Sicherheit, pharmazeutische Qualität und Wirksamkeit eines Impfstoffs zu stützen – und nichts anderes. Genehmigungen werden nur dann erteilt, wenn die Beweise überzeugend belegen, dass der Nutzen einer Impfung größer ist als die Risiken, die ein Impfstoff mit sich bringt.”
Autorin: Sonia Elijah
![]()
Am 05.10.22 erschienen auf: https://www.trialsitenews.com/a/a-further-investigation-into-the-leaked-ema-emails-confidential-pfizer-biontech-covid-19-vaccine-related-docs-5102039c
![]()
Übersetzung: Causalis (Hervorhebungen übernommen)
![]()
 Spendier mir 'nen Kaffee
Spendier mir 'nen Kaffee